Overview
The gsmr R-package implements the GSMR (Generalised Summary-data-based Mendelian Randomisation) method that uses GWAS summary statistics to test for a putative causal association between two phenotypes (e.g., a modifiable risk factor and a disease) based on a multi-SNP model (Zhu et al. 2018 Nature Communications). The R package is developed by Zhihong Zhu, Angli Xue, Zhili Zheng, Futao Zhang, and Jian Yang. Bug reports or questions: jian.yang@westlake.edu.cn.
Note: The GSMR method has also been implemented in the GCTA software (GCTA-GSMR)
Citation
Zhu, Z. et al. (2018) Causal associations between risk factors and common diseases inferred from GWAS summary data. Nature Communications, 9: 224.
Source code
Note: We have included a new HEIDI-outlier filtering method (as part of the GSMR analysis) since gsmr v1.0.7. The new HEIDI-outlier filtering method is currently under development and thus subject to further modifications. From the GSMR R package (>= version 1.0.8), we changed the default back to the original HEIDI-outlier method described in Zhu et al. (2018 Nature Communications) and added a temporary flag (‘gsmr2_beta’) to test the new method. The command to use this flag can be found in the tutorial below. The new HEIDI-outlier filtering method in gsmr (>= version 1.0.8) has been tested by extensive simulations and real data analyses. We will make a formal release of the method in our next GSMR paper.
Sample data are available in test_data.zip. This document has been integrated in the gsmr R-package, we can check it by the standard command “?function_name” in R.
Installation
The gsmr requires R >= 2.15, you can install it in R by:
# gsmr requires the R-package(s)
install.packages(c('survey'));
# install gsmr
install.packages("https://yanglab.westlake.edu.cn/software/gsmr/static/gsmr_1.1.1.tar.gz",repos=NULL,type="source")Update log
V1.1.1 (gmr_1.1.1.tar.gz PDF, 22 Jul. 2022): Fixed a bug of unused arguments error in V1.1.0.
V1.1.0 (gmr_1.1.0.tar.gz PDF, 13 Jun. 2022): Updated the multi-SNP-based HEIDI-outlier filtering method in gsmr2_beta.
V1.0.9 (gmr_1.0.9.tar.gz PDF, 18 Jun. 2019): Change the flag ‘gsmr_beta’ to ‘gsmr2_beta’.
V1.0.8 (gmr_1.0.8.tar.gz PDF, 21 Jan. 2019): Added a flag ‘gsmr_beta’ to use a testing version of the HEIDI-outlier method.
V1.0.7 (gmr_1.0.7.tar.gz PDF, 9 Oct. 2018): Added a multi-SNP-based HEIDI-outlier test in the HEIDI-outlier analysis.
V1.0.6 (gmr_1.0.6.tar.gz PDF, 23 Jan. 2018): Added a function to remove SNPs in high LD.
V1.0.5 (gmr_1.0.5.tar.gz PDF, 13 Dec. 2017): Improved the approximation of the sampling covariance matrix.
V1.0.4 (gsmr_1.0.4.tar.gz PDF, 6 Nov. 2017): Added the bi-directional GSMR analysis. The HEIDI-outlier analysis has been integrated in the GSMR analysis by default.
V1.0.3 (gsmr_1.0.3.tar.gz PDF, 12 Oct. 2017): Added more example data.
Removed the initial versions (8 Nov 2016).
Tutorial
The GSMR analysis only requires summary-level data from GWAS. Here is an example, where the risk factor (x) is LDL cholesterol (LDL-c) and the disease (y) is coronary artery disease (CAD). GWAS summary data for both LDL-c and CAD are available in the public domain (Global Lipids Genetics Consortium et al. 2013, Nature Genetics; Nikpay, M. et al. 2015, Nature Genetics).
1. Prepare data for GSMR analysis
1.1 Load the GWAS summary data
library("gsmr")
data("gsmr")
head(gsmr_data)## SNP a1 a2 a1_freq bzx bzx_se bzx_pval bzx_n bzy
## 1 rs10903129 A G 0.45001947 -0.0328 0.0037 3.030e-17 169920.0 0.008038
## 2 rs12748152 T C 0.08087758 0.0499 0.0066 3.209e-12 172987.5 0.013671
## 3 rs11206508 A G 0.14396988 0.0434 0.0055 2.256e-14 172239.0 0.030222
## 4 rs11206510 C T 0.19128911 -0.0831 0.0050 2.380e-53 172812.0 -0.074519
## 5 rs10788994 T C 0.18395430 0.0687 0.0049 8.867e-41 172941.9 0.038267
## 6 rs529787 G C 0.19713099 -0.0553 0.0052 8.746e-24 161969.0 0.001707
## bzy_se bzy_pval bzy_n
## 1 0.0092442 0.3845651000 184305
## 2 0.0185515 0.4611690000 184305
## 3 0.0141781 0.0330400000 184305
## 4 0.0133438 0.0000000234 184305
## 5 0.0118752 0.0012711000 184305
## 6 0.0135491 0.8997431000 184305dim(gsmr_data)## [1] 188 12This is the input format for the GSMR analysis. In this data set, there are 188 quasi-independent SNPs associated with LDL-c at a genome-wide significance level (i.e. p < 5e-8).
- SNP: the instrumental variable
- a1: the effect allele
- a2: the other allele
- a1_freq: frequency of a1
- bzx: the effect size of a1 on risk factor
- bzx_se: standard error of bzx
- bzx_pval: p value for bzx
- bzx_n: per-SNP sample size of GWAS for the risk factor
- bzy: the effect size of a1 on disease
- bzy_se: standard error of bzy
- bzy_pval: p value for bzy
- bzy_n: per-SNP sample size of GWAS for the disease
1.2 Estimate the LD correlation matrix
# Save the genetic variants and effect alleles in a text file using R
write.table(gsmr_data[,c(1,2)], "gsmr_example_snps.allele", col.names=F, row.names=F, quote=F)
# Extract the genotype data from a GWAS dataset using GCTA
gcta64 --bfile gsmr_example --extract gsmr_example_snps.allele --update-ref-allele gsmr_example_snps.allele --recode --out gsmr_exampleNote: the two steps above guarantee that the LD correlations are calculated based on the effect alleles of the SNPs.
# Estimate LD correlation matrix using R
snp_coeff_id = scan("gsmr_example.xmat.gz", what="", nlines=1)
snp_coeff = read.table("gsmr_example.xmat.gz", header=F, skip=2)# Match the SNP genotype data with the summary data
snp_id = Reduce(intersect, list(gsmr_data$SNP, snp_coeff_id))
gsmr_data = gsmr_data[match(snp_id, gsmr_data$SNP),]
snp_order = match(snp_id, snp_coeff_id)
snp_coeff_id = snp_coeff_id[snp_order]
snp_coeff = snp_coeff[, snp_order]
# Calculate the LD correlation matrix
ldrho = cor(snp_coeff)
# Check the size of the correlation matrix and double-check if the order of the SNPs in the LD correlation matrix is consistent with that in the GWAS summary data
colnames(ldrho) = rownames(ldrho) = snp_coeff_iddim(ldrho)## [1] 188 188# Show the first 5 rows and columns of the matrix
ldrho[1:5,1:5]## rs10903129 rs12748152 rs11206508 rs11206510 rs10788994
## rs10903129 1.000000000 -0.0045378845 0.008066621 -0.01372112 -0.0234447102
## rs12748152 -0.004537884 1.0000000000 -0.006687181 0.00445927 0.0003629201
## rs11206508 0.008066621 -0.0066871806 1.000000000 -0.21125757 0.0512593434
## rs11206510 -0.013721120 0.0044592696 -0.211257567 1.00000000 -0.1842706205
## rs10788994 -0.023444710 0.0003629201 0.051259343 -0.18427062 1.0000000000Note: all the analyses implemented in this R-package only require GWAS summary data (e.g., “gsmr_data”) and a LD correlation matrix computed from a reference sample (e.g., “ldrho”)
2. Standardization
This is an optional process. If the risk factor was not standardised in GWAS, the effect sizes can be scaled using the method below. Note that this process requires allele frequencies, z-statistics, and sample size. After scaling, bzx is interpreted as the per-allele effect of a SNP on the exposure in standard deviation units.
snpfreq = gsmr_data$a1_freq # allele frequencies of the SNPs
bzx = gsmr_data$bzx # effects of the instruments on risk factor
bzx_se = gsmr_data$bzx_se # standard errors of bzx
bzx_n = gsmr_data$bzx_n # GWAS sample size for the risk factor
std_zx = std_effect(snpfreq, bzx, bzx_se, bzx_n) # perform standardisation
gsmr_data$std_bzx = std_zx$b # standardized bzx
gsmr_data$std_bzx_se = std_zx$se # standardized bzx_se
head(gsmr_data)## SNP a1 a2 a1_freq bzx bzx_se bzx_pval bzx_n bzy
## 1 rs10903129 A G 0.45001947 -0.0328 0.0037 3.030e-17 169920.0 0.008038
## 2 rs12748152 T C 0.08087758 0.0499 0.0066 3.209e-12 172987.5 0.013671
## 3 rs11206508 A G 0.14396988 0.0434 0.0055 2.256e-14 172239.0 0.030222
## 4 rs11206510 C T 0.19128911 -0.0831 0.0050 2.380e-53 172812.0 -0.074519
## 5 rs10788994 T C 0.18395430 0.0687 0.0049 8.867e-41 172941.9 0.038267
## 6 rs529787 G C 0.19713099 -0.0553 0.0052 8.746e-24 161969.0 0.001707
## bzy_se bzy_pval bzy_n std_bzx std_bzx_se
## 1 0.0092442 0.3845651000 184305 -0.03055942 0.003447252
## 2 0.0185515 0.4611690000 184305 0.04713698 0.006234550
## 3 0.0141781 0.0330400000 184305 0.03829018 0.004852442
## 4 0.0133438 0.0000000234 184305 -0.07181919 0.004321251
## 5 0.0118752 0.0012711000 184305 0.06149455 0.004386074
## 6 0.0135491 0.8997431000 184305 -0.04695042 0.0044148683. GSMR analysis
This is the main analysis of this R-package. It uses SNPs associated with the risk factor (e.g., those with p < 5e-8) as the instruments variables to test for a putative causal effect of the risk factor on the disease. The analysis involves a step that uses the HEIDI-outlier filtering approach to remove SNPs that have effects on both the risk factor and the disease because of pleiotropy.
bzx = gsmr_data$std_bzx # SNP effects on the risk factor
bzx_se = gsmr_data$std_bzx_se # standard errors of bzx
bzx_pval = gsmr_data$bzx_pval # p-values for bzx
bzy = gsmr_data$bzy # SNP effects on the disease
bzy_se = gsmr_data$bzy_se # standard errors of bzy
bzy_pval = gsmr_data$bzy_pval # p-values for bzy
n_ref = 7703 # Sample size of the reference sample
gwas_thresh = 5e-8 # GWAS threshold to select SNPs as the instruments for the GSMR analysis
single_snp_heidi_thresh = 0.01 # p-value threshold for single-SNP-based HEIDI-outlier analysis
multi_snps_heidi_thresh = 0.01 # p-value threshold for multi-SNP-based HEIDI-outlier analysis
nsnps_thresh = 10 # the minimum number of instruments required for the GSMR analysis
heidi_outlier_flag = T # flag for HEIDI-outlier analysis
ld_r2_thresh = 0.05 # LD r2 threshold to remove SNPs in high LD
ld_fdr_thresh = 0.05 # FDR threshold to remove the chance correlations between the SNP instruments
gsmr2_beta = 0 # 0 - the original HEIDI-outlier method; 1 - the new HEIDI-outlier method that is currently under development
gsmr_results = gsmr(bzx, bzx_se, bzx_pval, bzy, bzy_se, bzy_pval, ldrho, snp_coeff_id, n_ref, heidi_outlier_flag, gwas_thresh, single_snp_heidi_thresh, multi_snps_heidi_thresh, nsnps_thresh, ld_r2_thresh, ld_fdr_thresh, gsmr2_beta) # GSMR analysis
filtered_index=gsmr_results$used_index
cat("The estimated effect of the exposure on outcome: ",gsmr_results$bxy)## The estimated effect of the exposure on outcome: 0.4322395cat("Standard error of bxy: ",gsmr_results$bxy_se)## Standard error of bxy: 0.02210985cat("P-value for bxy: ", gsmr_results$bxy_pval)## P-value for bxy: 4.15454e-85cat("Indexes of the SNPs used in the GSMR analysis: ", gsmr_results$used_index[1:5], "...")## Indexes of the SNPs used in the GSMR analysis: 1 2 3 5 6 ...cat("Number of SNPs with missing estimates in the summary data: ", length(gsmr_results$na_snps))## Number of SNPs with missing estimates in the summary data: 0cat("Number of non-significant SNPs: ", length(gsmr_results$weak_snps))## Number of non-significant SNPs: 39cat("Number of SNPs in high LD ( LD rsq >", ld_r2_thresh, "): ", length(gsmr_results$linkage_snps))## Number of SNPs in high LD ( LD rsq > 0.05 ): 5cat("Number of pleiotropic outliers: ", length(gsmr_results$pleio_snps))## Number of pleiotropic outliers: 94. Bi-directional GSMR analysis
The script below runs a bi-directional GSMR analysis, i.e., a forward GSMR analysis as described above and a reverse GSMR analysis that uses SNPs associated with the disease (e.g., those with p < 5e-8) as the instrumental variables to test for a putative causal effect of the disease on risk factor.
gsmr_results = bi_gsmr(bzx, bzx_se, bzx_pval, bzy, bzy_se, bzy_pval, ldrho, snp_coeff_id, n_ref, heidi_outlier_flag, gwas_thresh, single_snp_heidi_thresh, multi_snps_heidi_thresh, nsnps_thresh, ld_r2_thresh, ld_fdr_thresh, gsmr2_beta) # GSMR analysis
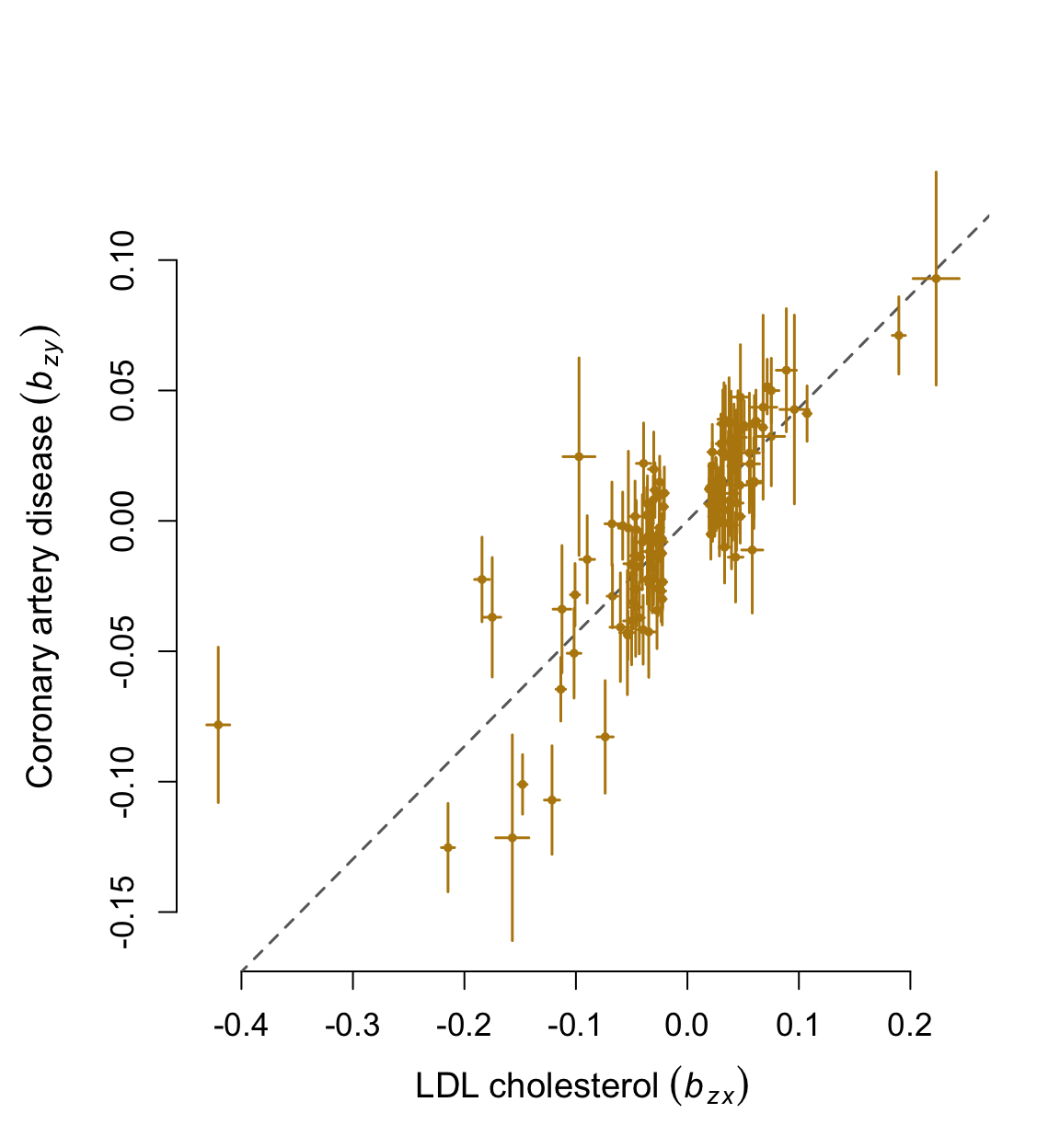
cat("Effect of risk factor on disease: ",gsmr_results$forward_bxy)## Effect of risk factor on disease: 0.4322395cat("Standard error of bxy in the forward-GSMR analysis: ",gsmr_results$forward_bxy_se)## Standard error of bxy in the forward-GSMR analysis: 0.02210985cat("P-value of bxy in the forward-GSMR analysis: ", gsmr_results$forward_bxy_pval)## P-value of bxy in the forward-GSMR analysis: 4.15454e-85cat("Effect of disease on risk factor: ",gsmr_results$reverse_bxy)## Effect of disease on risk factor: -0.02739421cat("Standard error of bxy in the reverse-GSMR analysis: ",gsmr_results$reverse_bxy_se)## Standard error of bxy in the reverse-GSMR analysis: 0.009551025cat("P-value of bxy in the reverse-GSMR analysis: ", gsmr_results$reverse_bxy_pval)## P-value of bxy in the reverse-GSMR analysis: 0.0041281985. Visualization
effect_col = colors()[75]
vals = c(bzx[filtered_index]-bzx_se[filtered_index], bzx[filtered_index]+bzx_se[filtered_index])
xmin = min(vals); xmax = max(vals)
vals = c(bzy[filtered_index]-bzy_se[filtered_index], bzy[filtered_index]+bzy_se[filtered_index])
ymin = min(vals); ymax = max(vals)
par(mar=c(5,5,4,2))
plot(bzx[filtered_index], bzy[filtered_index], pch=20, cex=0.8, bty="n", cex.axis=1.1, cex.lab=1.2,
col=effect_col, xlim=c(xmin, xmax), ylim=c(ymin, ymax),
xlab=expression(LDL~cholesterol~(italic(b[zx]))),
ylab=expression(Coronary~artery~disease~(italic(b[zy]))))
abline(0, gsmr_results$forward_bxy, lwd=1.5, lty=2, col="dim grey")
nsnps = length(bzx[filtered_index])
for( i in 1:nsnps ) {
# x axis
xstart = bzx[filtered_index [i]] - bzx_se[filtered_index[i]]; xend = bzx[filtered_index[i]] + bzx_se[filtered_index[i]]
ystart = bzy[filtered_index[i]]; yend = bzy[filtered_index[i]]
segments(xstart, ystart, xend, yend, lwd=1.5, col=effect_col)
# y axis
xstart = bzx[filtered_index[i]]; xend = bzx[filtered_index[i]]
ystart = bzy[filtered_index[i]] - bzy_se[filtered_index[i]]; yend = bzy[filtered_index[i]] + bzy_se[filtered_index[i]]
segments(xstart, ystart, xend, yend, lwd=1.5, col=effect_col)
}
Package Document
bi_gsmr
The bi-directional GSMR analysis is composed of a forward and a reverse GSMR analysis that uses SNPs associated with the disease (e.g., those with < 5e-8) as the instrumental variables to test for a putative causal effect of the disease on the risk factor.
Usage
bi_gsmr(bzx, bzx_se, bzx_pval, bzy, bzy_se, bzy_pval, ldrho, snpid, heidi_outlier_flag=T, gwas_thresh=5e-8, single_snp_heidi_thresh=0.01, multi_snps_heidi_thresh=0.01, nsnps_thresh=10, ld_r2_thresh=0.05, ld_fdr_thresh=0.05, gsmr2_beta=0)
Arguments
bzx |
vector, SNP effects on risk factor |
bzx_se |
vector, standard errors of bzx |
bzx_pval |
vector, p values for bzx |
bzy |
vector, SNP effects on disease |
bzy_se |
vector, standard errors of bzy |
bzy_pval |
vector, p values for bzy |
ldrho |
LD correlation matrix of the SNPs |
snpid |
instrumental variables (i.e., SNPs) |
n_ref |
sample size of the reference sample |
heidi_outlier_flag |
HEIDI-outlier filtering analysis |
gwas_thresh |
threshold p-value to select SNP instruments from GWAS for risk factor |
single_snp_heidi_thresh |
p-value threshold for the single-SNP-based HEIDI-outlier filtering analysis |
multi_snps_heidi_thresh |
p-value threshold for multi-SNP-based HEIDI-outlier filtering analysis |
nsnps_thresh |
the minimum number of SNP instruments required for the GSMR analysis (we do not recommend users to set this number smaller than 10) |
ld_r2_thresh |
LD r2 threshold to remove SNPs in high LD |
ld_fdr_thresh |
An FDR threshold to remove chance correlations between the SNP instruments |
gsmr2_beta |
GSMR2 beta version, including a new HEIDI-outlier filtering method (used as part of the GSMR analysis) that is currently under development and subject to future modifications, 0 - the original HEIDI-outlier filtering method, 1 - the new HEIDI-outlier filtering method |
Value
The estimated causative effect of risk factor on disease (forward_bxy), the corresponding standard error (forward_bxy_se), p-value (forward_bxy_pval) and SNP index (forward_index), andi the estimated causative effect of disease on risk factor (reverse_bxy), the corresponding standard error (reverse_bxy_se), p-value (reverse_bxy_pval), SNP index (reverse_index), SNPs with missing values, with non-significant p-values and those in LD.
Examples
data("gsmr")
gsmr_result = bi_gsmr(gsmr_data$bzx, gsmr_data$bzx_se, gsmr_data$bzx_pval, gsmr_data$bzy, gsmr_data$bzy_se, gsmr_data$bzy_pval, ldrho, gsmr_data$SNP, n_ref, T, 5e-8, 0.01, 0.01, 10, 0.05, 0.05, 0)
gsmr
GSMR (Generalised Summary-data-based Mendelian Randomisation) is a flexible and powerful Mendelian Randomization (MR) method that utilises GWAS summary statistics to test for a causal association between two phenotypes (e.g., a modifiable risk factor and a disease) based on a multi-SNP model.
Usage
gsmr(bzx, bzx_se, bzx_pval, bzy, bzy_se, ldrho, snpid, heidi_outlier_flag=T, gwas_thresh=5e-8, single_snp_heidi_thresh=0.01, multi_snps_heidi_thresh=0.01, nsnps_thresh=10, ld_r2_thresh=0.05, ld_fdr_thresh=0.05, gsmr2_beta=0)
Arguments
bzx |
vector, SNP effects on risk factor |
bzx_se |
vector, standard errors of bzx |
bzx_pval |
vector, p values for bzx |
bzy |
vector, SNP effects on disease |
bzy_se |
vector, standard errors of bzy |
ldrho |
LD correlation matrix of the SNPs |
snpid |
Instrument variables (i.e., SNPs) |
n_ref |
sample size of the reference sample |
heidi_outlier_flag |
HEIDI-outlier filtering analysis |
gwas_thresh |
threshold p-value to select SNP instruments from GWAS for risk factor |
nsnps_thresh |
the minimum number of SNP instruments required for the GSMR analysis (we do not recommend users to set this number smaller than 10) |
ld_r2_thresh |
LD r2 threshold to remove SNPs in high LD |
ld_fdr_thresh |
An FDR threshold to remove chance correlations between the SNP instruments |
gsmr2_beta |
GSMR2 beta version, including a new HEIDI-outlier method (used in a GSMR analysis) that is currently under development and subject to future changes, 0 - the original HEIDI-outlier method, 1 - the new HEIDI-outlier method |
single_snp_heidi_thresh |
p-value threshold for single-SNP-based HEIDI-outlier analysis |
multi_snps_heidi_thresh |
p-value threshold for multi-SNP-based HEIDI-outlier analysis |
Value
Estimate of causative effect of risk factor on disease (bxy), the corresponding standard error (bxy_se), p-value (bxy_pval), SNP index (used_index), SNPs with missing values, with non-significant p-values and those in LD.
Examples
data("gsmr")
gsmr_result = gsmr(gsmr_data$bzx, gsmr_data$bzx_se, gsmr_data$bzx_pval, gsmr_data$bzy, gsmr_data$bzy_se, ldrho, gsmr_data$SNP, n_ref, T, 5e-8, 0.01, 0.01, 10, 0.1, 0.05, 0)
std_effect
Estimating standardized SNP effect from z-statistic, allele frequency, and sample size
Usage
std_effect(snp_freq, b, se, n)
Arguments
snp_freq |
vector, allele frequencies |
b |
vector, estimated SNP effects on the risk factor |
se |
vector, standard errors of b |
n |
vector, per-SNP sample sizes of GWAS for the risk factor |
Value
Standardised effect (b) and the corresponding standard error (se)
Examples
data("gsmr")
std_effects = std_effect(gsmr_data$a1_freq, gsmr_data$bzx, gsmr_data$bzx_se, gsmr_data$bzx_n)